Research Overview
The Greenberg laboratory has a long-standing interest in uncovering the mechanisms by which experience-driven neural activity shapes the developing mammalian nervous system. Beginning the mid-1980s with the appreciation that growth factors and neurotransmitters trigger the rapid transcription of the Fos gene, the laboratory has focused on elucidating the nature and role of neuronal transcriptional programs triggered in response to extracellular stimuli.
To this end, we have worked to characterize the various signaling pathways that convey neurotrophin and calcium-dependent signals from distal synapses to the neuronal nucleus and sought to elucidate the role of neuronal activity-induced transcriptional responses as mediators of neural circuit plasticity, including the exploration of characterized effects on neuronal survival, dendritic remodeling, synaptic strength, and synapse development and elimination. Over time, these studies have expanded to encompass the investigation of additional neuronal kinase-dependent signaling networks. Given the strong links between these processes and various human disorders of cognitive function, we continually seek to exploit these molecular insights to advance understanding of the etiology and treatment of clinically relevant neurological conditions. Specific areas of current investigation within the laboratory include…
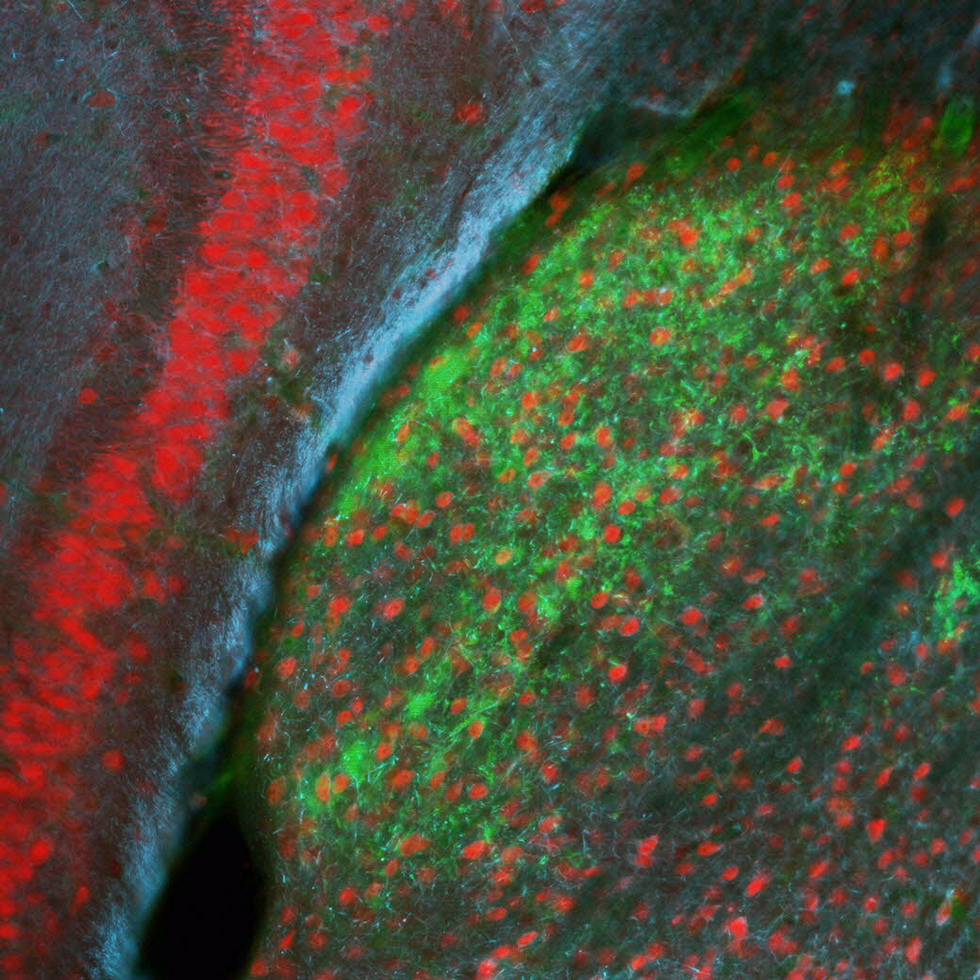

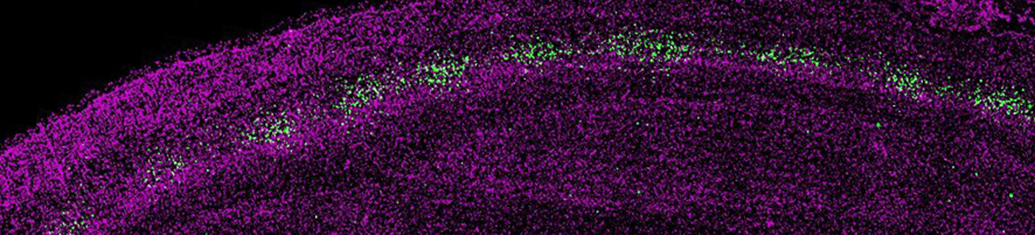
Sensory-driven circuit development
Sensory experience influences neural circuit assembly and refinement in large part by driving the expression of specialized cohorts of genes in neurons of the developing brain. These important activity-dependent gene programs are highly cell type- and brain region-specific and have thus been challenging to study in vivo. To overcome these challenges, we have begun characterizing the effects of these gene programs within the context of specific neural circuits. Using a combination of molecular genetics, high-throughput sequencing, and electrophysiological techniques, we are currently characterizing the transcriptional programs that are specifically activated in relay neurons of the dorsal lateral geniculate nucleus of the thalamus, cells that receive sensory information about the visual world from the retinal ganglion cells of the eye, to determine how these gene programs contribute to sensory-driven circuit refinement. We also continue to investigate how activity-dependent gene programs regulate the number and plasticity of inhibitory synapses within neural circuits.
Bloodgood, B.L., Sharma, N., Browne, H.A., Trepman, A.Z. & Greenberg, M.E. (2013). The activity- dependent transcription factor NPAS4 regulates domain-specific inhibition. Nature 503(7474): 121-125. PMCID: PMC4169177
Spiegel, I., Mardinly, A.R., Gabel, H.W., Bazinet, J.E., Couch, C.H., Tzeng, C.P., Harmin, D.A. & Greenberg, M.E. (2014). Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 157(5): 1216-1229. PMCID: PMC4089405
Mardinly, A.R., Spiegel I., Patrizi A., Centofante E., Bazinet J.E., Tzeng C.P., Mandel-Brehm C., Harmin D.A., Adesnik H., Fagiolini M., Greenberg M.E. (2016). Sensory experience regulates cortical inhibition by inducing IGF1 in VIP neurons. Nature. 17;531(7594):371-5. PMCID: PMC4823817
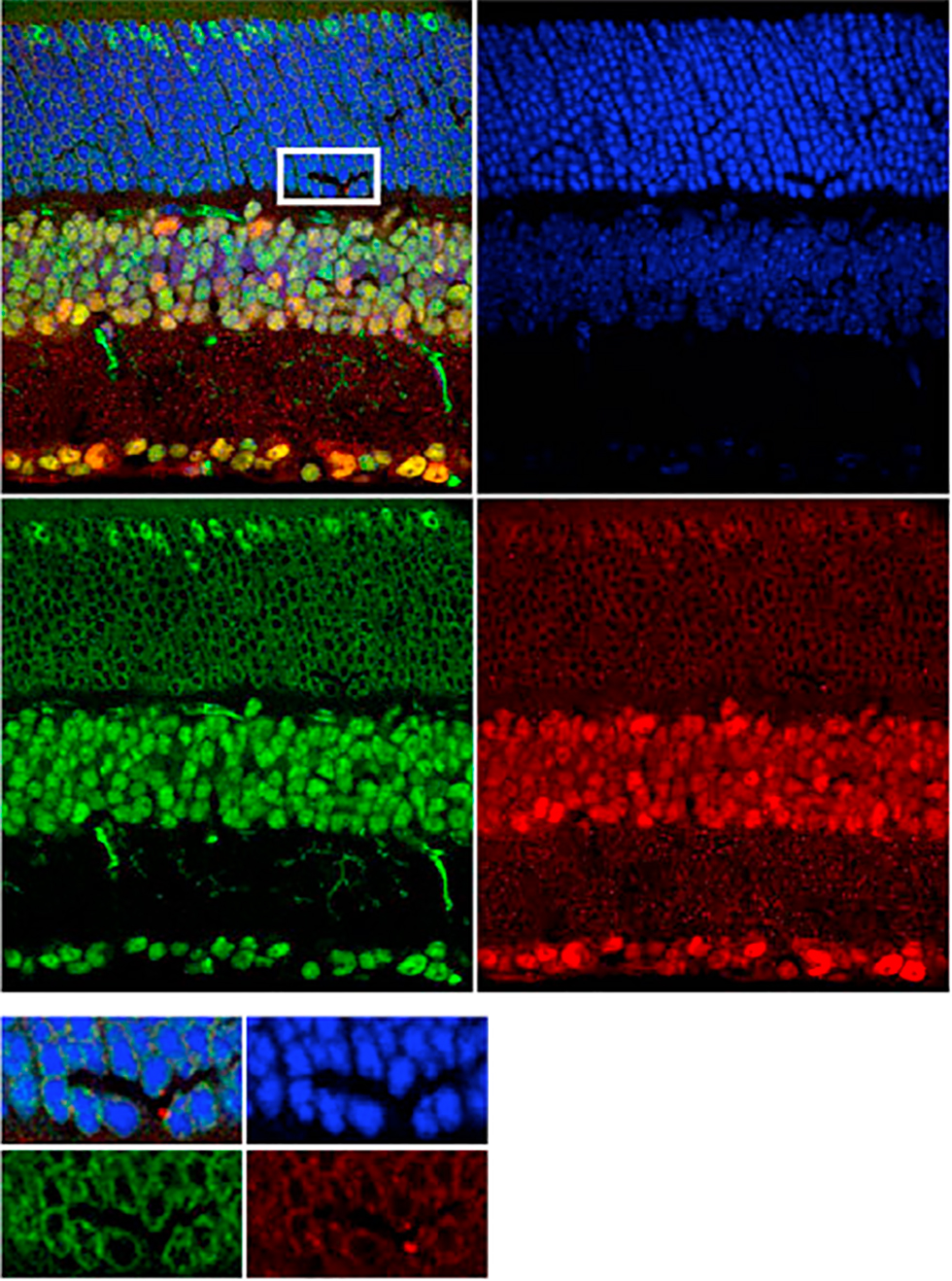
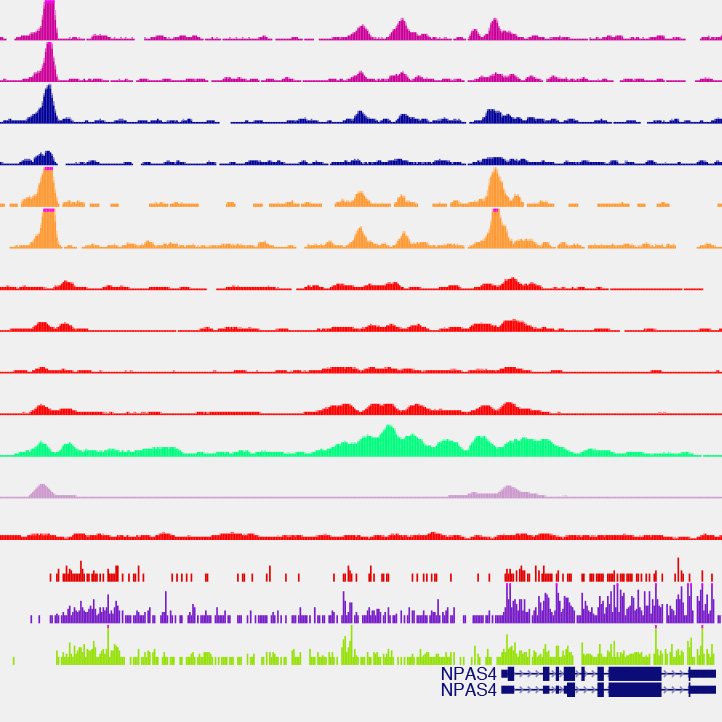
Distal enhancer elements in neuronal activity-dependent transcriptional responses
We are interested in understanding how enhancers regulate expression of cell type-specific stimulus-responsive genes. We are characterizing neuronal subtype-specific experience-dependent gene expression programs in an effort to understand how different neuronal subtypes are differentially regulated by experience and the manner in which distal enhancer elements contribute to this regulation. We hope to uncover additional transcriptional regulators that drive the cell type-specific activation of these enhancers, with the long-term goal of understanding how genetic variation within these regulatory elements shapes transcriptional outputs and modulates neuronal activity-dependent phenotypes. In addition, we continue to study the mechanistic role played by specific transcriptional regulators at stimulus-responsive enhancers. In this regard, we have found that the canonical immediate-early gene transcription factor AP-1 (Fos, FosB, Jun, JunB) binds almost exclusively to enhancer elements and is required for their function in many cases. Using genomic and proteomic approaches, we hope to uncover how AP-1 functions at enhancers to promote the transcription of cell type-specific stimulus-responsive genes.
Kim, T.K., Hemberg, M., Gray, J.M., Costa, A.M., Bear, D.M., Wu, J., Harmin, D.A., Laptewicz, M., Barbara-Haley, K., Kuersten, S., Markenscoff-Papadimitriou, E., Kuhl, D., Bito, H., Worley, P.F., Kreiman, G. & Greenberg, M.E. (2010). Widespread transcription at neuronal-activity regulated enhancers. Nature 465(7295): 182-187. PMCID: PMC3020079
Andzelm, M.M., Cherry, T.J., Harmin, D.A., Boeke, A.C., Lee, C., Hemberg, M., Pawlyk, B., Malik, A.N., Flavell, S.W., Sandberg, M.A., Raviola, E. & Greenberg, M.E. (2015). MEF2D drives photoreceptor development through a genome-wide competition for tissue-specific enhancers. Neuron 86(1): 247-263. PMCID: PMC4393375
Malik, A.N., Vierbuchen, T., Hemberg, M., Rubin, A.A., Ling, E., Couch, C.H., Stroud, H., Spiegel, I., Farh, K.K., Harmin, D.A. & Greenberg, M.E. (2014). Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nature Neuroscience 17(10): 1330-1339. PMCID: PMC4297619
Human molecular neurobiology
Most of what we know about mammalian neurobiology comes from invaluable studies in rodent models. However, we are increasingly aware of the need for non-rodent model systems to achieve a full understanding of human neurodevelopment and disease. To begin to address human-specific aspects of neuronal activity-dependent signaling, we are exploring the activity-dependent epigenetic, transcriptional, and post-transcriptional responses of human neurons using multiple platforms. Human induced pluripotent stem cells (iPSCs) are particularly useful for these studies as they can be differentiated into a variety of neuronal populations that can then be analyzed through genome-wide approaches.
Ataman B*, Boulting GL*, Harmin DA, Yang MG, Baker-Salisbury M, Yap EL, Malik AN, Mei K, Rubin AA, Spiegel I, Durresi E, Sharma N, Hu LS, Pletikos M, Griffith EC, Partlow JN, Stevens CR, Adli M, Chahrour M, Sestan N, Walsh CA, Berezovskii VK, Livingstone MS, Greenberg ME. Evolution of Osteocrin as an activity-regulated factor in the primate brain. Nature. 2016 Nov 9;539(7628):242-247. PMID: 27830782
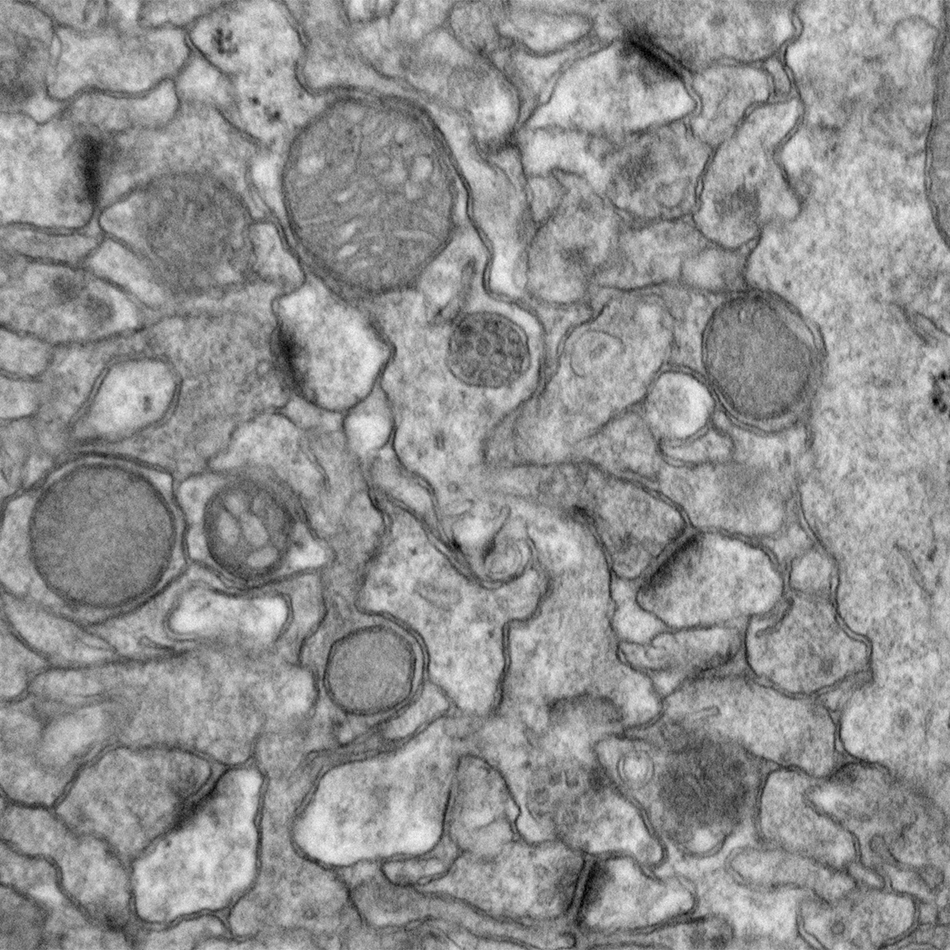
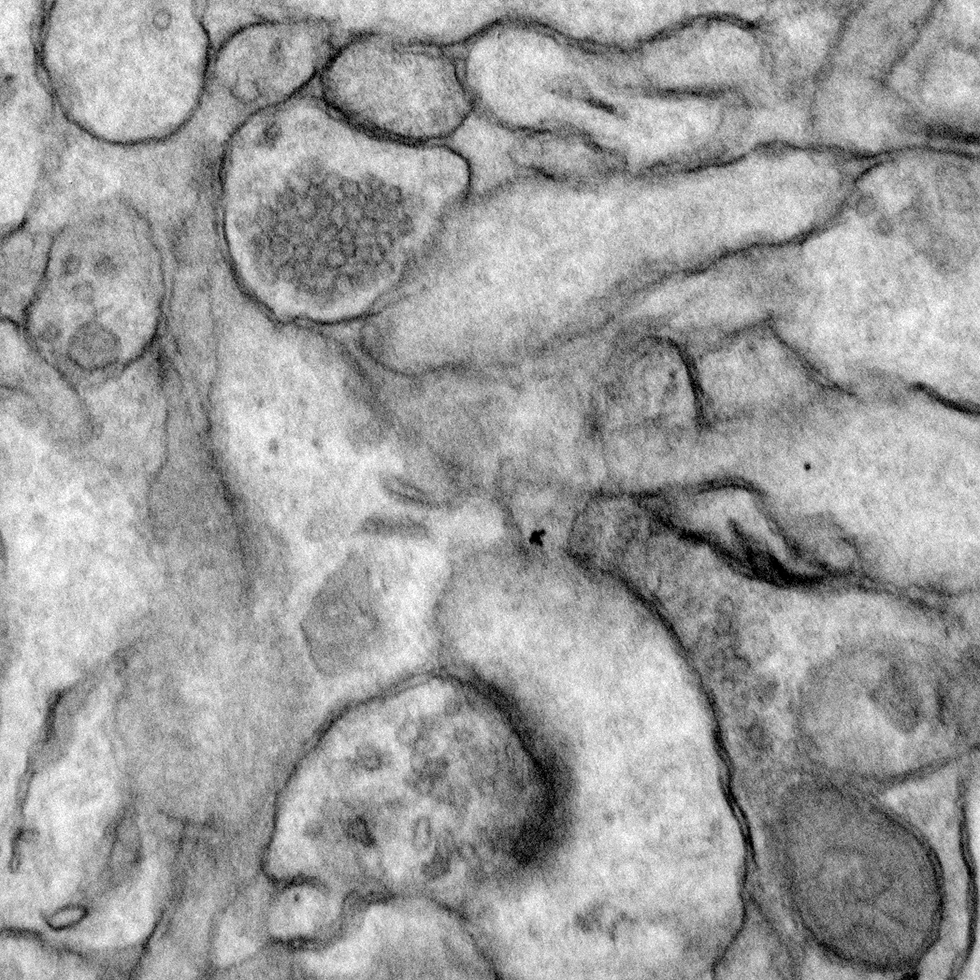
Eph tyrosine kinase signaling in neural development
The Eph subfamily of receptor tyrosine kinases mediates multiple aspects of neural development, including long-range axonal pathfinding, synaptogenesis, and synaptic plasticity. Moreover, disruption of Eph function has been linked to neurological disorders such as autism and Alzheimer’s disease. Building on our earlier studies demonstrating that EphBs play key role in promoting excitatory synapse formation in the developing nervous system, we have conducted an extended investigation of EphB-dependent signaling pathways, elucidating important novel mediators of Eph signaling. Current studies focus on Ephexin5, a critical negative regulator of EphB signaling, exploring its role both in normal neural development as well as in cases of infantile epilepsy.
Margolis, S.S., Salogiannis, J., Lipton, D.M., Mandel-Brehm, C., Wills, Z.P., Mardinly, A.R., Hu, L., Greer, P.L., Bikoff, J.B., Ho, H.Y., Soskis, M.J., Sahin, M. & Greenberg, M.E. (2010). EphB-mediated degradation of the RhoA GEF Epheinx5 relieves a developmental brake on excitatory synapse formation. Cell 143(3): 442-455. PMCID: PMC2967209
Soskis, M.J., Ho, H.Y., Bloodgood, B.L., Robichaux, M.A., Malik, A.N., Ataman, B., Rubin, A.A., Zieg, J., Zhang, C., Shokat, K.M., Sharma, N., Cowan, C.W. & Greenberg, M.E. (2012). A chemical genetic approach reveals distinct EphB signaling mechanisms during brain development. Nature Neuroscience 15(12): 1645-1654. PMCID: PMC3509236
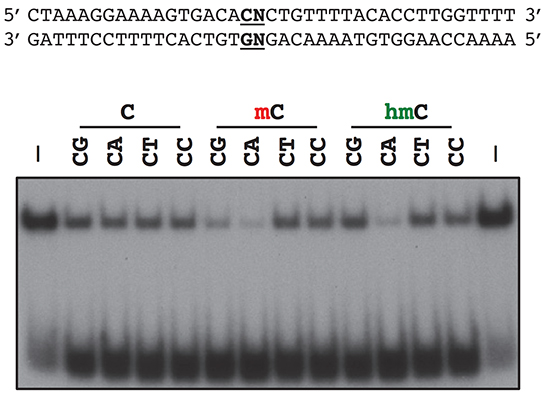
MECP2 function and Rett Syndrome:
Mutations in MeCP2, a methyl-DNA-binding protein that functions as a regulator of gene expression, are a major cause of Rett syndrome (RTT), an X-linked progressive neurological disorder associated with profound cognitive impairment in girls and women. While loss of MeCP2 function in post-mitotic neurons is sufficient to give rise to RTT phenotypes, the precise function of MeCP2 in neurons and how the disruption of these mechanisms gives rise to RTT remain unclear. We are using a combination of genomic, biochemical, electrophysiological, and behavioral approaches to study the molecular function of MeCP2, with a specific focus on its role in neuronal activity and development. In this regard, we have identified a number of post-translational modifications of MeCP2 that are regulated by neuronal stimuli as well as distinctive genomic features that are important for MeCP2 action. We hope to translate these new molecular insights back to patients through novel pharmacologic and genetic manipulations in preclinical animal models of Rett syndrome.
Ebert, D.H., Gabel, H.W., Robinson, N.D., Kastan, N.R., Hu, L.S., Cohen, S., Navarro, A.J., Lyst, M.J., Ekiert, R., Bird, A.P. & Greenberg, M.E. (2013). Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499(7458): 341-351. PMCID: PMC3922283
Gabel, H.W., Kinde, B., Stroud, H., Gilbert, C.S., Harmin, D.A., Kastan, N.R., Hemberg, M., Ebert, D.H. & Greenberg, M.E. (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522(7554): 89-93. PMCID: PMC4480648